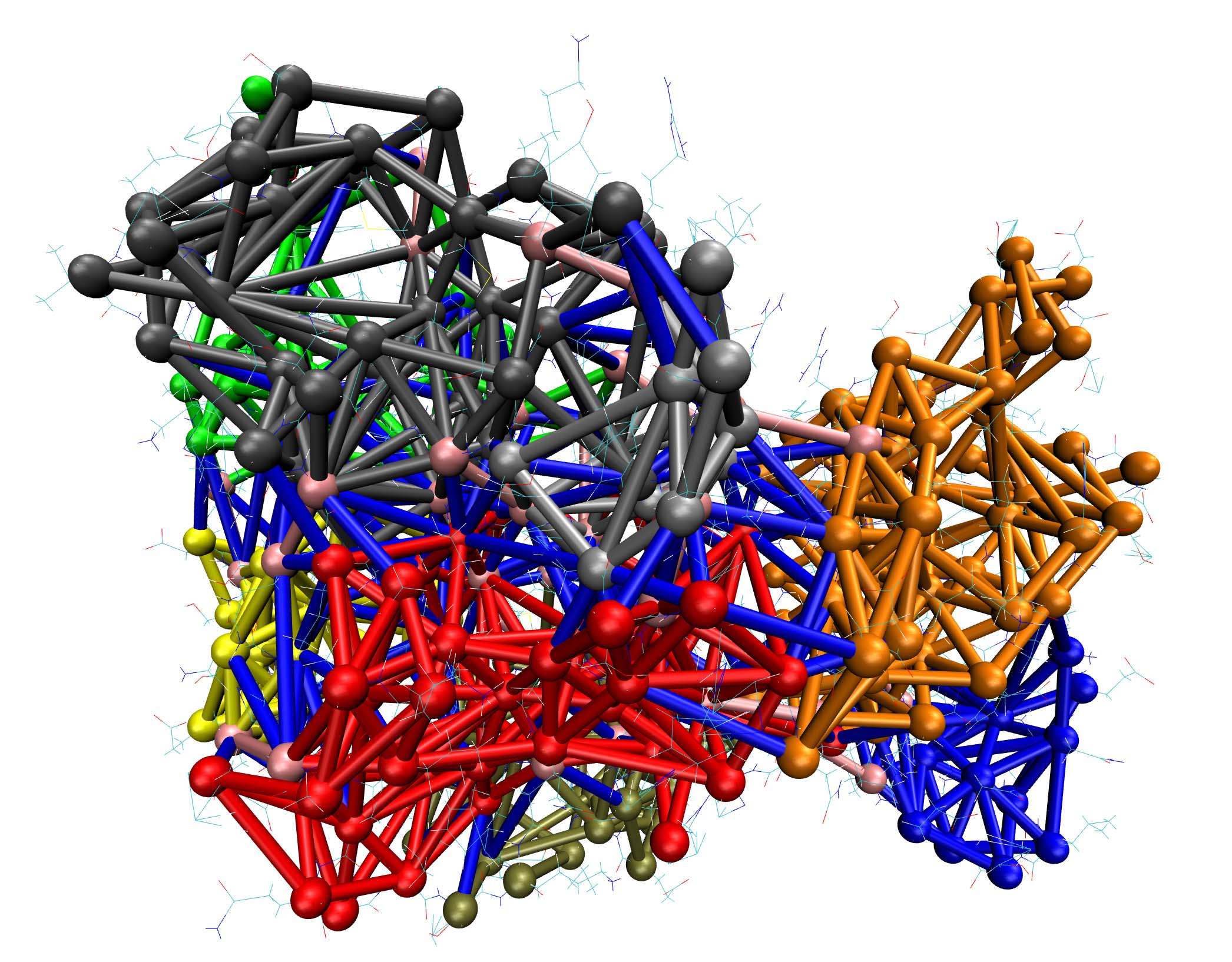
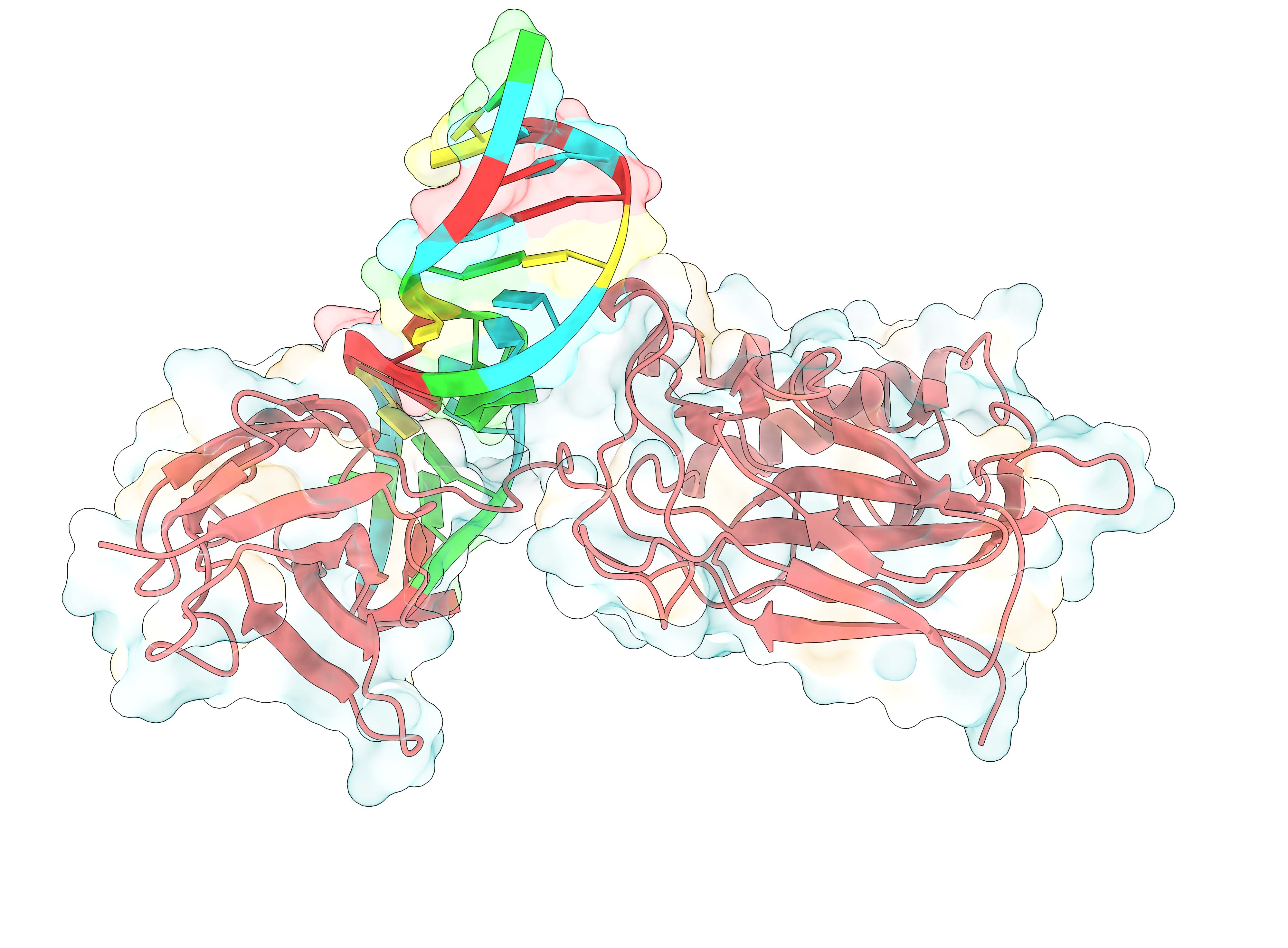
We developed interpretable machine learning-based network technologies to study protein-protein/protein-RNA binding dynamics, enhancing sample efficiency and magnifying interaction changes. The strategy has been successfully applied to study multi-body interactions in complex biomolecular systems. We successfully developed an interpretable machine learning-based network RNet. RNetsite employs a machine learning-based network decomposition algorithm to predict RNA binding sites by analyzing the local and global network properties. Our study shows that RNetsite outperforms existing methods, achieving precision values as high as 0.701 on TE18 and 0.788 on RB9 tests. We also developed RNetdyn, a distance-based dynamic graph algorithm, to characterize the consequences of the interface dynamical behavior upon inhibitor binding. The simulation testing of competitive inhibitors indicates that RNetdyn outperforms the traditional method by 30% (Briefings in Bioinformatics, 2024. Highlighted story). Tcf1- and Lef1-deficient CD8+ T cells exhibit histone hyperacetylation, ascribed to intrinsic histone deacetylase (HDAC) activity in Tcf1 and Lef1. Mutation of five conserved amino acids in the Tcf1 HDAC domain diminishes HDAC activity and the ability to suppress CD4+ lineage genes in CD8+ T cells (Nature Immunology, 2016. Highlighted story).